


Time:2015-10-26 11:54:29
药审新政将给行业带来5大巨变
8月18日,国务院对外发布《关于改革药品医疗器械审评审批制度的意见》(以下简称“意见”),业内期盼已久的药品审评审批改革的大幕正式拉开。药审新政将深刻改变我国医药产业格局,未来行业将发生巨大变化。
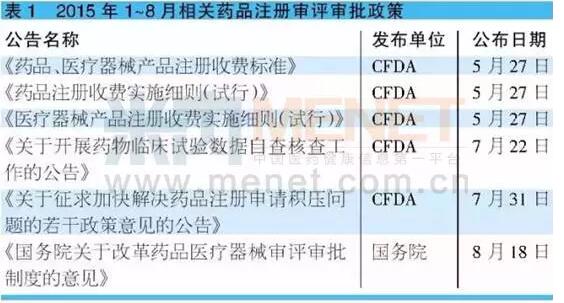
巨变一:药品质量将大幅度提高
国家食品药品监管总局副局长吴浈表示:“国务院关于改革药品医疗器械审评审批制度的意见,核心就是提高药品的质量,通过改革来促进医药行业产业的结构调整和转型升级,实现上市产品的有效性和安全性、质量可控性,能够达到国际先进水平,满足公众的用药需求,这就是这次起草这样一个改革方案的背景。”
本次改革目标上包括提高药品审批质量、提高仿制药水平、鼓励创新等内容,提高审批标准解决的增量部分,一致性评价工作则解决已经上市使用的存量药品的质量提高问题。未来随着行业“优胜劣汰”,对那些质量差、研发能力弱的企业是“利空”,但对优秀的医药企业,对国内医药行业来说都是好事。
巨变二:审评积压将得到很快解决
在中国,新药审批速度太慢是各方诟病已久的话题。国内一款药品从前期试验到上市批准需要8年到10年的过程。吴浈列了一组数字:现在在审的药品有2.1万个品种,其中90%是仿制药。再进一步细化,其中8个品种有100多家企业在申报;23个品种有50-99家在申报;89个品种有20-49家企业申报。
针对这些问题,《意见》明确提出,要建立科学、高效的药品医疗器械审评审批体系,严格控制市场供大于求药品的审批。吴浈表示,2016年初步消除积压;2017年实现年度进出平衡;2018年将按时限完成审评。
具体政策方面体现在以下四个方面:
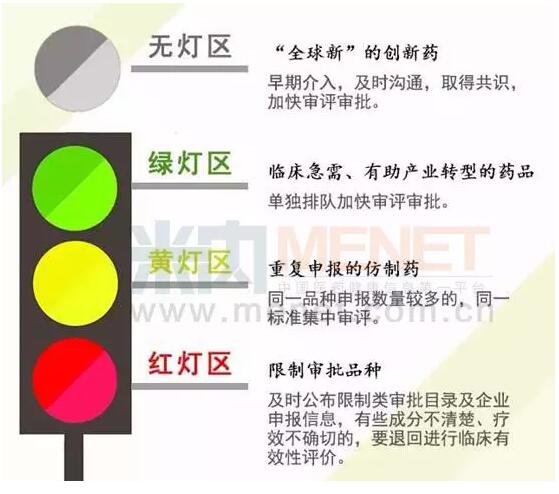
第一,将“全球新”的创新药归入“无灯”区,早期介入,及时沟通,取得共识,加快审评审批;
第二,临床急需、有助于产业转型的药品归入“绿灯”区,实行单独排队加快审评审批;
第三,重复申报仿制药归入“黄灯”区,同一品种申报数量较多的实行同一标准集中审评;具有审评价值但存在瑕疵的,退回企业补充资料后继续审评;
第四,将限制审批品种归入“红灯”区,要及时公布限制类审批目录,及时公布企业申报信息,减少企业重复和不合理申报。有些成分不清楚、疗效不确切的,要退回进行临床有效性评价。
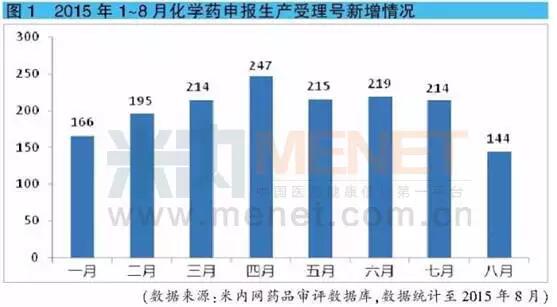
据米内网药品审评数据库统计,2015年1~7月,每月新增化学药申报生产的受理号数量均维持在165~250之间,主要以仿制药申报为主。而8月份新增化学药申报生产受理号数量出现明显下滑,仅为144个,较7月份下降32%。可见,政策叠加带来的影响最先在药品申报中体现。
变化三:一致性评价将纵深推进

如果说提高审批标准解决的是增量部分,一致性评价工作就是解决已经上市使用的存量药品的质量提高问题。
《关于改革药品医疗器械审评审批制度的意见》将提高仿制药质量,加快仿制药质量一致性评价工作列为改革五大目标之一。力争2018年底前完成国家基本药物口服制剂与参比制剂质量一致性评价。就业界关注的参比制剂问题,新政明确由CFDA征询专家意见后确定,可选择原研药品,也可选择国际公认的同种药品。无参比制剂的,由企业进行临床有效性试验。规定期限内未通过的不予再注册。
44号文发布当日,国新办少见地就药审改革举行新闻发布会,接下来,质量一致性评价工作将呈现蹄疾步稳、纵深推进的态势。
变化四:鼓励创制新药

创新是解决临床问题的根本手段,是企业提高产品质量的基础,是实现医药产业转型升级的重要支撑。《意见》对创新药的审评审批给予诸多方便,旨在加快创新药上市节奏。《意见》提出,对创新药实行特殊审评审批制度;加快审评审批防治艾滋病、恶性肿瘤、重大传染病、罕见病等疾病的创新药,列入国家科技重大专项和国家重点研发计划的药品,转移到境内生产的创新药和儿童用药,以及使用先进制剂技术、创新治疗手段、具有明显治疗优势的创新药。
同时,开展药品上市许可持有人制度试点。允许药品研发机构和科研人员申请注册新药,在转让给企业生产时,只进行生产企业现场工艺核查和产品检验,不再重复进行药品技术审评。
从本次药审改革文件由国务院层面下发就可以看出,国家对于我国新药研发的重视程度已经上升到战略层面。这意味着中国制药创新将迎来新的发展阶段。
变化五:3类新药消失
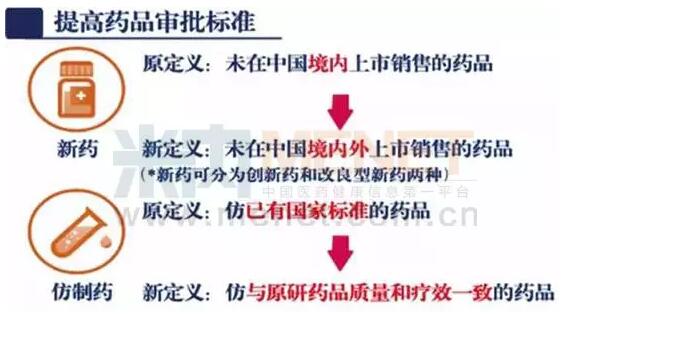
重新定义和分类新药是国务院印发的《关于改革药品医疗器械审评审批制度的意见》(以下简称《意见》)中最大的改变之一。文件谈到的首要任务就是提高药品审批标准,并将药品分为新药和仿制药,而新药又分为创新药和改良型新药。《意见》提出,将新药由现行的“未曾在中国境内上市销售的药品”调整为“未在中国境内外上市销售的药品”。根据物质基础的原创性和新颖性,将新药分为创新药和改良型新药。
目前,详细的新药注册分类配套方案仍在酝酿之中,但管理部门此次对药品分类重新定调,无疑将导致传统分类中的3类药物进入仿制药范畴,从而对该类药物的注册上市、市场开拓产生巨大影响。
以往热衷申报3.1类新药的企业受此次新药定义调整的影响最为明显,这类药品的新药地位不再,企业只能等仿制,并且做到质量和临床疗效一致才有可能获批上市。
10月27~29日,第27届全国医药经济信息发布会在泰州举行,国家食品药品监督管理总局副局长吴浈将现场讲解《中国药审改革的顶层设计与制度创新》,中国食品药品检定研究院副院长张志军将解读《中国仿制药一致性评价实施关键点》,敬请期待。(米内网)