


Time:2015-09-05 11:24:43
发达国家向非发达国家发射的“三枚烟雾弹”
自2013年11月在《中国医药工业杂志》发表“仿制药研发中有关物质研究思路之我见”后,收到大量同仁来电来函交流。积累一年多心得,撰写此续篇,希冀能将某些观点阐述得更为清楚明了,进而为我国仿制药研发中杂质研究思路与控制策略提供一种更为客观理性的认知,为科学厘清杂质对于药物临床使用的价值与意义呈上绵薄之力,为正确认清本行业高科技的核心与实质献计献策,为将我们的有限资源用到研发关键之处指明方向。
1. 从宏观上解读杂质
1.1. 杂质与药物不良反应的关系
……
1.2. 杂质与药物质量控制的关系
……
1.3. 发达国家向非发达国家施放的“烟雾弹”
国内对杂质研究之所以“着力猛攻、一网打尽”,还与盲目迷信国外文献资料有关。由于制药行业的利润实在太过丰厚,所以吾等千万不要幼稚地以为“老外们会很傻/很天真地和盘托出”,相反原研企业会千方百计地在仿制药研发与申报的征途中设置一些障碍或施放一些“烟雾弹”来迷惑我们……。举例如下:
1.3.1. 第一枚烟雾弹 —— 杂质
秉承制剂质量标准仅关注降解杂质的原则,在前些年的国外药典和进口质量标准中很多制剂品种无有关物质检查项,这是科学合理的:经研究当“原料药制成0天制剂”和“0天制剂在加速试验6个月/长期试验6个月/货架期内”,这两个环节中杂质均无变化(不是没有、是无变化),便可在制剂质量标准中无需制订有关物质检查项,从而实现发达国家早在多年前就开始提倡和践行的“绿色检验”理念——为减少环境污染与试剂排放,检验工作应尽可能做到事半功倍、一针见血。
但近些年,这些标准中却开始制订并收载大量杂质,且有愈演愈烈之态势,乍看起来甚是“高标准。严要求”,但本人认为这是在施放“烟雾弹”、目的是引仿制者误入歧途。解读如下案例:
某一普通口服片剂进口质量标准(规格为1.0mg~0.125mg)中收载了12个已知杂质,并采用了极为复杂的色谱系统进行测定,还引入校正因子逐一准确计算,并制定了每一杂质限度。秉承上述原则,该药物是不可能在效期内有12个降解杂质产生的。果然,经对数批货架期内原研制剂样品予以检测或取1批样品进行加速试验6个月测试,绝大部分都是未检出(报告限以下)、仅有1~2个杂质在报告限以上、鉴定限以下,根本无需研究。可令人遗憾的是:国内众多同仁将进口质量标准/国外药典奉为“圭臬”、当作“圣旨”,花费大量金钱与资源去设法获得那12个杂质,最终经研究,在仿制原料药和仿制制剂中均是未检出,因我国合成能力极强,完全有能力将原料药中杂质全部去除;再加上主成分的自身稳定性很好、制成制剂后杂质也不会有变化。
针对该品种,本人推荐研发思路如下:直接采用既有质量标准中的色谱条件、无需验证,第一步/测定三批原研制剂样品,并经对杂质谱剖析、无含量不断增加杂质;第二步/测定三批规模化生产的仿制原料药与仿制制剂样品,无制剂鉴定限以上杂质,且无含量不断增加杂质;第三步/根据以上研究结果,最终在制剂的质量标准中无需制订有关物质检查项,原料药质量标准也仅是粗略地制订单杂和总杂即可,根本无需制订那么多杂质。甚至采取更为大胆的作法——由于主成分规格很小,且口服,每日最多3片,故杂质与残留溶剂均无需研究,只需在申报材料中阐明:经推算这些物质的每日最高摄入量均小于每日临床安全摄入限度即可,从而做到专业的最高境界——有所为有所不为。
还有一进口外用涂抹皮肤的乳膏剂质量标准,主成分规格依然很小、却制订了8个已知杂质。本人认为这些均是原研企业对仿制者施放的烟雾弹,目的就是阻碍仿制进程、迷惑研发方向的表现。
至于如何看待国外药典原料药项下罗列的众多杂质,且毫无保留地给出了结构式、花钱还可购买来(价格甚是不菲),那更是仁者见仁、智者见智。但此处笔者想提醒的是:目前全世界绝大多数原料药均已由吾等发展中国家生产后出口至ICH国家(把雾霾和污染留给了我们自己),发达国家要做的:是将这些“已把杂质几乎抠没”的原料药,经过特殊的、保密处理后(例如重结晶后制成目标晶型、粒度分布、颗粒形状、比表面能、晶格能、固有溶出速率等特性)制成制剂、再销往全世界获取高额利润,当然质量绝对毋庸置疑。
1.3.2. 第二枚烟雾弹 —— 溶出度试验
各剂型均有关键性评价指标,在ICH Q6文件(规范:新原料药和新药制剂的测试方法和认可标准:化学物质)和世界卫生组织药品标准专家委员会第43次技术报告一书中均有相关阐述。
发达国家针对这些关键性评价指标,要么隐含(仅在严格保密的、企业内控质量标准中才有),要么即便公开、也大多为宽松的试验参数与限度范围。如对于口服固体制剂,最为核心的是溶出度试验。本人曾接触过上百个原研制剂多条溶出曲线剖析测定结果,发现相当一部分国外药典和进口质量标准中的试验参数都极为宽松、不具区分力(如采用高转速、高浓度表面活性剂、或是高溶解度的溶出介质)。盖因该试验太重要,连发达国家在制订ICH Q系列质量管理文件时都未有专门的指导原则进行阐述,唯《日本溶出曲线数据库》收载的实验条件是真实的、最具区分力的条件。
这里需阐明:日本自上世纪九十年代起就十分重视体外溶出度试验,因日方很早就意识到生物等效性(BE)试验由于仅采用年轻男性作为受试者有其局限性(故BE试验不是“金标准”),在无法采用各种人群大标本试验的前提下,只能通过对体外溶出度试验的严格要求来促使仿制制剂内在品质“无限趋近”原研制剂。为此,日本在1997年出版的《仿制药生物等效性试验指导原则》中就已有了详尽的溶出度试验研究要求,并占据了一半篇幅。同时,迄今为止日本官方已建立起700多个品种的原研制剂四条溶出曲线数据库,并予以了全面的真实公开,且该项工作还在不断进行中……。
而欧美的公开资料中则大多宣称:体内生物利用度试验才具有最终决定性,不太强调体外溶出度试验。但通过解读他们公开文献中的溶出度试验参数,发现多数不具区分力、甚至干脆不公开(如英国药典收载的制剂很少,甚至有些难溶性口服固体制剂竟然没有溶出度试验,缓控释制剂更是一个品种也无释放度试验)的作法说明:这是一种韬光养晦、瞒天过海的表现。如我们按此种不具区分力的实验条件去研制仿制制剂,则多半会出现“部分仿制药(主要是那些有制剂难度的品种:难溶性口服固体制剂、肠溶制剂、缓控释制剂、pH值依赖型制剂、治疗窗狭窄药物制剂等)对于部分患者(尤中老年人)安全无效、吃了白吃”的结果。不深谙溶出度评价,就不知努力的方向与差距所在,因溶出度试验才是撬动制剂内在品质不断提升的“那根杠杆”,只有掌握科学客观的评价法,才能做出与原研制剂内在品质无限趋近的高品质仿制药来,这将充分体现“标准就是生产力”的精神。
1.3.3. 第三枚烟雾弹 —— 潜在基因毒性杂质
确实发生过类似事件,但绝对是“极小概率”。我国撷取后营造出的学术氛围就犹如发现了一大“宝藏”,提高至“报告限以上杂质就应考虑,甚至潜在杂质也要考虑”;而“一旦潜在、就无穷无尽了”。近三年,针对该类杂质开展了轰轰烈烈的研究,花费巨资购买了大量进口高精尖检测设备,而最终样品检测结果很多都是“未检出”。
其实,只要观测仿制原料药的“起始物、副产物、试剂、配位体、催化剂”等这些物质是否具备文献报道的“基因毒性杂质官能团结构式”,并针对规模化生产的仿制原料药三批样品中检出的鉴定限(0.10%)以上特有杂质(即原研制剂中不存在的)是否具备这些结构式,如具备再进行相应研究便是,切忌“大惊小怪”、犹如“惊弓之鸟”。
现今,随着科学发展人们认知到:连药物自身都会给人体带来致癌毒性。所以,强烈建议我国药品审评中心(CDE)的审判员和药物研发者均无需过于谨小慎微、患得患失。
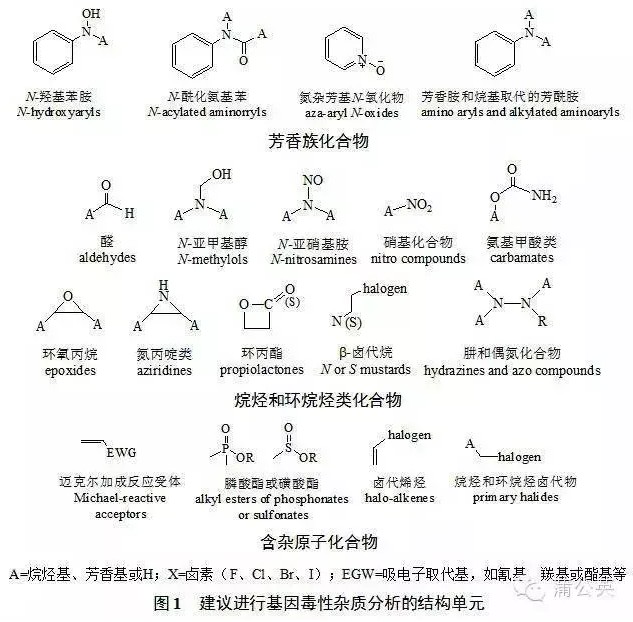
A为烷烃基、芳香基或H;X为卤素,包括F、Cl、Br、I;EGW为吸电子取代基,如氰基、羰基或酯基等
图1 具备以上结构单元的杂质、建议进行基因毒性杂质的分析和说明
1.3.4. “其他烟雾弹”
今后一定还会有其他“烟雾弹”发射过来,如近期兴起的“金属杂质研究”等。
1.3.5 无独有偶
1.3.6. 应对策略
首先我们应理解这种行为无可厚非,这是由本行业的特殊性所决定的(“N高” —— 高投入、高利润、高附加值、高端制造业)。我们作为发展中国家,在仿制过程中,要具备“公开的就不是高科技、看不到的才是高科技”之辩证唯物观点。具体建议如下:
(1) 针对进口质量标准 相关药检机构在进行复核时,应要求外企提供详尽的检测法验证资料,即应着重审阅质量标准中的各试验参数是如何建立的、关注其逻辑推导过程,是经怎样的研究内容与试验结果确定的,这才是审核重点与核心;如未尽详细,应要求对方持续提供。如溶出度试验,为何申报中制订了100rpm高转速和该溶出介质?是否研究过50和75转溶出行为以及其他介质的溶出情况后综合考虑确定?是否有故意放宽、让自我产品永远合格的嫌疑。又比如有关物质,为何订入这些多个杂质和这么低的限度,制订的依据是否充分等等。
(2) 针对公开的各国药典与文献资料 切忌照搬照抄、盲目迷信。CDE早在2003年就已提出“仿产品不是仿标准”的指导思想,就是希冀众人在具体研制时能独立思考、冷静分析。遗憾的是;很多同仁认为该宗旨中的“标准”仅是指国内质量标准、不包括国外标准/文献,导致被这些标准迷惑、误入歧途。本人从事药物分析工作17年,深感:发达国家的分析检测水平很一般、有时甚至拙劣,他们强在药剂、尤工业药剂学上。
(3) 总之,无论如何都应获取至少三批原研制剂,深度剖析其关键质量特性参数后再来指引自我仿制药的开发和制订质量标准等事宜,这才是研发的根本出发点,也唯有如此才能做到“知己知彼、百战不殆”。